
-
Koji
This is a great video on making sake.
https://www.youtube.com/watch?v=UA83cK1Xouk
Mad respect for this guy. Every artesenal distiller knows that felling of sweating your ass off and wondering what am I doing only to taste your product and be satisfied you have created something new.
GNS Continuous Still?@grim said: Jesus, 14 years ago…
We were all doing so much experimentation at that stage. most of us didn't have the science of it like you guys, a lot was 'what if'.
Everything from equipment, to methods, to recipes.
@Smaug's thumper pumper was out there too.....
@olddog brought the bubble plate column to the hobby world not long before we all started StillDragon and doing all our product development.
Was a cool time to be part of the hobby.
GNS Continuous Still?Jesus, 14 years ago…
GNS Continuous Still?Growing on the previous picture...
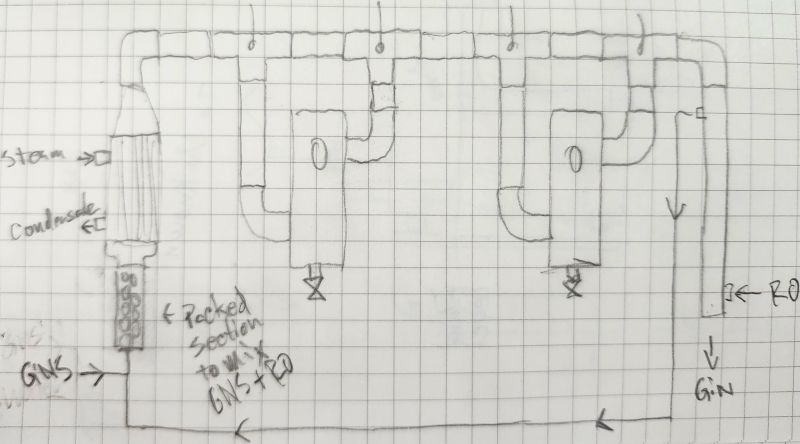
What if you were to do a thumper of sorts with an overflow below the vapor basket with all your masserated botanicals? This would be a highly concentrated maceration. You don't need the thumper for proof but as a way to get some of the more full flavors you don't get with vapor basket.
 GNS Continuous Still?
GNS Continuous Still?Not at all wrong.
I tried this once using 100% neutral, on accident. Had the water valve off, so I was pumping only neutral. Flavor profile was very different. I let it run through for a while for the hell of it. Then, not sure why, I decided to just try running water alone, and just running steam through.
The end result was very similar to the normal run. Each individual component was not. The steam distilled botanicals ran white like milk. But as soon as the distillate hit the very-high-proof first runnings, it went clear again.
Toyed around a few times after that, but when I ran a vapor proof that was identical to my batch distilled product proof (before dilution), the end result was identical.
The best I could posit was, the same amount of alcohol vapor and water vapor is passing through the botanicals. The only difference is that they are constant through the run, and not starting high/ending low/vice-versa.
Assuming you are getting full extraction of your botanicals, why would the end result differ? it all comes down to grams/liter of extract and oils per finished bottle. Keep those ratios identical, it won't matter.
You could get very fancy with this setup, and vary the ratio of water/ethanol through the run, in a manner similar to a step mash in brewing. (5 minutes at 190 proof, 5 minutes at 150 proof, 17 minutes at 130 proof, 8 minutes at 110 proof, etc, etc). You could actually get better consistency doing this than you ever could with a batch still.
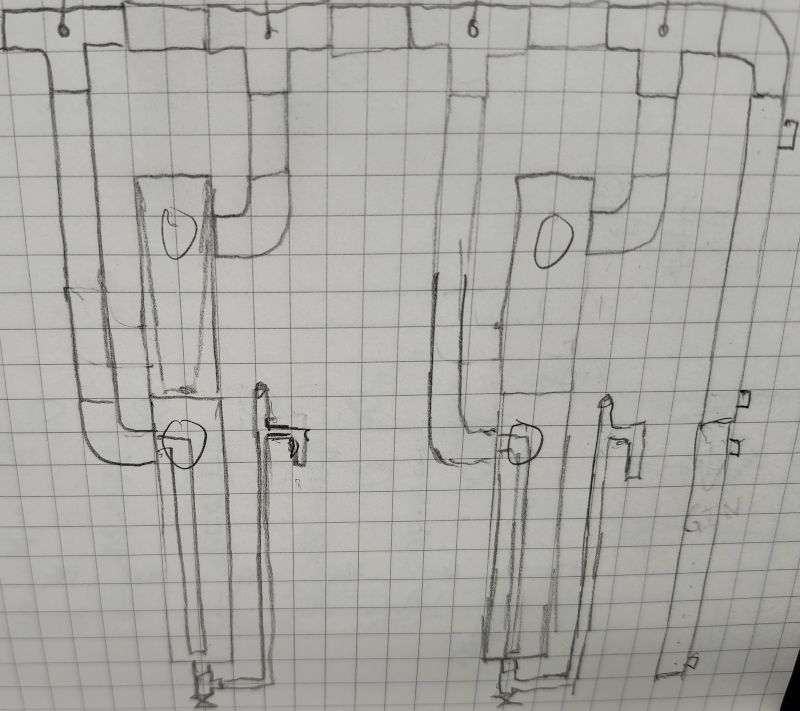
GNS Continuous Still?@grim is this more or less that you had in mind for the continuous gin still?
 Rotovap
RotovapThe vacuum rating would also need to apply to the collection vessel, and any associated piping.
Though the piping is probably the least worrisome part of this.
Column diameters do need to be upsized for vacuum use, as the vapor speeds are far higher, and flooding becomes a major issue.
Realistically, you'd probably need a 12" or 18" column on a 380l tank to keep vapor speed in check. Or settle with pot-still only.
RotovapThe minute you pull vacuum on a vessel in a commercial setting, you enter the same territory as running a high pressure vessel (in the US anyway, though would imagine similar in Europe). This means your vacuum vessels are going to need to carry a certification (ASME in the US), a nameplate with design limits, and usually annual testing that your local government, and probably insurance company, will want. Generally if a tank is going to be under partial vacuum, it's prudent to design it to operate under Full Vacuum (usually indicated as FV on the nameplate), to avoid catastrophic damage if full vacuum is accidentally pulled, which is usually pretty easy.
Just because a tank can support # psi pressure, does not mean it can support the same differential as vacuum. There is no rule that works to convert a pressure rating to a vacuum rating.
Good example is a soda can, which is designed to hold anywhere from 90-120 psi pressure, but pull even a small vacuum and it fails catastrophically.
Might a tank that isn't rated work once or twice? Sure, maybe. 50 times? Maybe, maybe not.
Cue obligatory vacuum catastrophe video:
RotovapThe kettles are rated for 15 psi.
EDIT: 15 psi positive pressure. I have no idea if there is a negative pressure rating. Our factory does not test for negative pressure.
Stripping Run --> Spirit run: How to minimise wastage?20:1 is used on HD
TōtōGNS Continuous Still?@Bolverk said: Croz, I think that's how Smaug is recommending doing it. Standard CM upper configuration but the dephleg is tuned to only let heads past and then condensed in the product condenser.
Yessir. Definitely helps when calculating actual processing speed.
Steve Todee in Bastrop has settled in on 2L of heads collection for every 100 gallons of beer as an example. Not saying this is "thee" be all concluded ratio btw. It's going to depend on everyone's sensory awareness perception.
GNS Continuous Still?@Smaug, I see what you're saying. What I'm thinking is a cross between the GNS continuous and the LM function of the spirit side of your continuous... like a boka but you're taking off the heads at the LM port and your good spirit out of the bottom of the column.
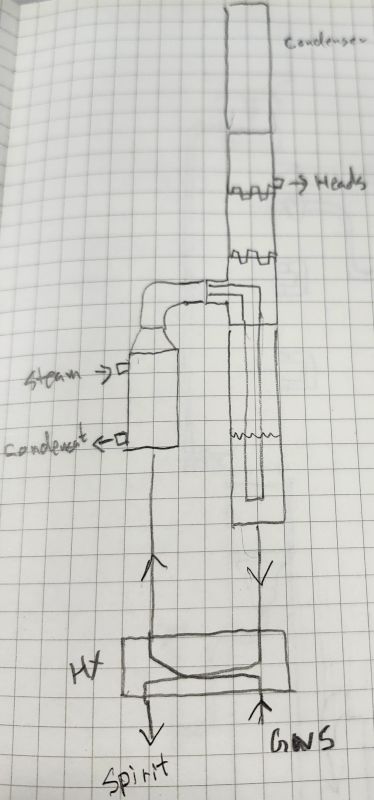 GNS Continuous Still?
GNS Continuous Still?@Smaug said: So far the feedback in doing this has shown to further assist in making a more precise on the fly heads cut.
And having said that, if the 4 plate spirit column is for bourbon only, we'll be putting a plate disabler system on. The dedicated bourbon only still only needs 2 plates if you want to go straight into the barrel at an ideal barreling proof. 4 plates allows for finished spirit to come off in the high 150s. The reflux enriches the lower plates too much.
GNS Continuous Still?Yeah, I'm trying to work out a few things too including:
Reboiler HX appears to be a 4" x 750mm shell in tube
column
- Diameter? looks to be about 8"
- Length? about 1m?
- Plate type: sieve vs bubblecaps?
- plate count: 4?
- how do the level sensors control the operation of the unit?
condensor
- looks like a 4" x 1m long
- How does it work to take off heads? Are they drawn off via the vent? As there's a flame arrestor I don't think so as it'd also need a condenser for the heads take off
- Is the line from the bottom of the condenser to close to the bottom of the column the condensed hearts?
Pumps
- What would be the specs of the 2 pumps used for feed in and product out?
- Do they run continuous or does the level sensor(s) turn it on/off?
- I think they'd need to be ATEX/IECEX = $$$
- Could double diaphram pneumatic pumps be used instead of electric?
At 2gpm that comes to 462lph! 96% GNS has a boiling point around 78C so the Preheat HX & reboiler don't have as much work to do compared to running a 10% wash which needs to get to about 93C. So trying to work out what I could do with my available power
GNS Continuous Still?That makes sense, thank you
I guess what I dont understand is how they are removing the heads vapor. It appears to be venting out via the fire arrestor, but how does that work? Is the temp in the condenser kept at say 173°f to allow for the etoh the condense but the heads to remain a vapor?
GNS Continuous Still?The operating principal is pretty simple:
- There is a single pass boiler (not reboiler), which vaporizes near 100% of the feed, injecting that vapor into the column, which operates like. thumper. There are no plates in the column.
- Vapor moves up into the condenser, gets condensed, and flows BACK INTO THE "column". You can see the drain located about one quarter/third from the left on the top condenser. It flows to the bottom of the column.
- Column liquid volume is controlled by the level sensor, which operates the discharge valve. There are two pumps, a feed pump, and a discharge pump.
- There is a temperature probe on the vapor vent from the top condenser (port at the top, far end). There is a PID (visible ABB unit) that controls the water flow through that top condenser using the vapor vent temp as process input. The set point controls the amount of input that's removed as vapor.
- The discharge pump drains from the bottom of the column, through the heat exchanger, out.
Everything in the "distillation" stream is product, there are no bottoms (as this is GNS), the only thing removed is a very small amount of "heads" as vapor.
GNS Continuous Still?Photos of the Vendome GNS still in real life: Vendome Continuous GNS Still (New!!) @ ADI Forums
Stripping Run --> Spirit run: How to minimise wastage?@richard said: Seems excessive. Say 125mm diameter x 30 = 3.75m. Normally a minimum of 1.2m packed section is sufficient plus RC etc. on top. I could relate to this if it were plates at pitches of say 100mm and 30 plates were the requirement.
Thanks. Yeah I can't remember what that rule of thumb was.
Stripping Run --> Spirit run: How to minimise wastage?@Smaug said: No sir. The diameter to height ratio for columns with random packing materials.
Seems excessive. Say 125mm diameter x 30 = 3.75m. Normally a minimum of 1.2m packed section is sufficient plus RC etc. on top. I could relate to this if it were plates at pitches of say 100mm and 30 plates were the requirement.
Fats in the Pool?Both methanol and ethanol will react with long chain fatty acids found in meat or vegetables to form the precursors of diesel fuel (FAME/FAEE) - basically just long chain esters. They don't all come across as heads products, the longer chain stuff smears across hearts, tails, and beyond.
I could see locking up some methanol in longer-chain esters, and being able to cut that out as tails.
The protein stuff, that stumps me. I need to study aldehyde chemistry more, aldehydes will react with proteins and amino acids.
Stuff like:
Understanding interactions among aldehyde compounds and porcine myofibrillar proteins by spectroscopy and molecular dynamics simulations
porcine myofibrillar proteins = pork meat
Among flavour compounds, aldehydes have a significant role in flavour development of meat and meat products [9], [10]. Some studies describe the influences of aldehyde characteristics (such as structure and concentration) on their interactions with protein, and some investigations concluded that aldehydes react with amino acid residues of proteins via irreversible covalent binding [20], [21]. Other studies reported that hydrophobic interactions were the predominant force responsible for the interactions among proteins and aldehydes [22]. Based on these studies, both reversible and irreversible binding modes appear to be the major contributors to the interaction among proteins and aldehydes, depending on the specific chemistry and structure of the interacting molecules. Nevertheless, information on the conformational changes that occur in MPs upon aldehyde binding remains limited, and the mechanisms of aldehydes binding to MPs remain unexplored.


